Kazlauskas Lab
The overall goal of the Kazlauskas Lab is to elucidate the effect of diabetes (DM) on the retinal vasculature. The resulting conceptual advances will guide development of new therapeutic approaches to prevent patients with DM from developing diabetic retinopathy (DR), and improve current options to treat patients who have already developed DR. Several areas of research in the Kazlauskas Lab are summarized below; please see our recent publications for additional information:
View select publications on PubMed
Ongoing Research in the Kazlauskas Lab Heading link
-
A novel strategy to prevent diabetic retinopathy
 Fig 1: RDR involves enhanced mitochondrial flux. The deleterious impact of chronically elevated HG includes increased mitochondrial oxidative stress and dysfunction. Retinal endothelial cells keep mitochondrial oxidative stress in check by eliminating the damaged mitochondria via mitophagy. A matched increase in mitochondrial biogenesis sustains mitochondrial homeostasis. Thus, enhanced mitochondrial flux in the face of HG is the core of RDR (resilience to DR).
Fig 1: RDR involves enhanced mitochondrial flux. The deleterious impact of chronically elevated HG includes increased mitochondrial oxidative stress and dysfunction. Retinal endothelial cells keep mitochondrial oxidative stress in check by eliminating the damaged mitochondria via mitophagy. A matched increase in mitochondrial biogenesis sustains mitochondrial homeostasis. Thus, enhanced mitochondrial flux in the face of HG is the core of RDR (resilience to DR).Diabetic retinopathy (DR) is the most common microvascular complication of diabetes mellitus (DM) and the leading cause of blindness among working-age individuals. DR eventually develops in the vast majority of patients with DM and is the most-feared complication of DM, perhaps because loss of vision profoundly impacts an individual’s quality of life.
Available approaches to treat the vision-threatening forms of DR (proliferative diabetic retinopathy (PDR) and macular edema) are not ideal. Not all patients respond to anti-VEGF, steroids increase the risk of glaucoma, and laser permanently damages the retina. Furthermore, these treatments address symptoms as opposed to the root cause of vascular dysfunction, which is hyperglycemia (HG)-driven death of cells within the retinal capillaries.
In light of the issues accompanying treatment options for patients who have developed DR, it is imperative to prevent the manifestation of this complication. The opportunity to prevent DR is especially promising because at-risk patients are readily identifiable (those who have developed DM), and there is a long delay between the onset of DM and manifestation of DR; 21.3 years of DM before developing PDR. Furthermore, there are effective strategies to delay DR. Despite these favorable conditions for prevention, 26% of patients with DM have DR, indicating that available approaches to prevent DR are not effective for over 1/4 of this patient population.
Current therapeutic strategies to prevent and treat DR are based on our understanding of DR pathogenesis. For instance, curbing blood glucose to prevent DR arose from the realization that HG damages the retina and is a major risk factor associated with DR. Similarly, anti-VEGF-based therapy was guided by the discovery of VEGF, which drives vascular dysfunction (leakage and angiogenesis) that compromises vision. New insights into the mechanism of DR pathogenesis will enable development of additional therapeutic approaches. Our discovery that the pathogenesis of DR involves deterioration of an endogenous system that protects from the deleterious effects of DM constitutes a major advance in our understanding of DR pathogenesis. This discovery is the scientific premise of this project seeking to develop a novel therapeutic strategy to prevent DR.
We are uniquely poised to execute this project because of our recently published conceptual and technical advances in the resilience to DR (RDR) arena. We discovered that RDR exists in mouse models of both T1D and T2D DM/DR, and we developed bioassays and biomarkers of RDR. Furthermore, we established in vitro models of RDR and used them to identify governors of RDR (Fig 1). Serendipitously, genetic, pharmacological and nutraceutical approaches to either activate or antagonize the governors of RDR have been developed and extensively characterized. These tools enable and ensure the feasibility of this project.
-
A bioinformatics-based approach to identify new therapeutic approaches and targets for PDR
The therapeutic efficacy of anti-VEGF (vascular endothelial growth factor) in patients with diabetic retinopathy (DR) demonstrates the central role of VEGF in the pathogenesis of this vascular disease. Not all patients experience adequate benefit from anti-VEGF, and even those who do, can suffer detrimental side effects of this therapeutic option. The overall goal of this project is to identify new therapeutic approaches and targets for patients who have developed vision threatening forms of diabetic retinopathy such as proliferative diabetic retinopathy (PDR) diabetic macular edema (DME).
The standard of care for patients that have advanced to end-stage PDR is surgery to remove pathological blood vessels. The availability of such clinical specimens constitutes an opportunity to investigate the transcriptional profile within pathological blood vessels in patients. We embraced this opportunity by assembling a team to consent patients, collect surgical specimens, isolate the CD31+ cells from the pathological vessels, and determine their gene expression profile using RNA sequencing. To date, we generated high-quality PDR patient endothelial transcriptomes from the CD31+ cells from ten end-stage PDR patients. This project involves mining these transcriptomes to define the molecular signature of PDR within the endothelium. The molecular signature will then be used to establish novel classes of therapeutic targets, identify alternative targets for anti-VEGF therapy, and generate pre-clinical data for the best targets.
Join Us Heading link
Ambitious individuals who are interested in joining the Kazlauskas Lab should send their CV and names of three references to ak20@uic.edu.
Applicants must be M.D. or Ph.D. candidates in biological science, cell and molecular biology, ophthalmology or related fields with 0-2 years of experience.
Past Contributions Heading link
-
Details
The discovery of kinases that phosphorylate proteins on tyrosine residues and their association with proliferation of cells captured the attention of both basic and translational researchers. As a postdoctoral fellow in Jonathan Cooper’s lab I focused on the receptor for platelet-derived growth factor (PDGF). I discovered that tyrosine autophosphorylation of the PDGFR not only enhanced this receptor’s intrinsic kinase activity, but created docking sites for SH2 domain-containing proteins. The PDGFR autophosphorylates at multiple tyrosine residues, which, together with the surrounding amino acids, constitute a binding site for a specific a SH2 domain-containing protein. Other groups focusing on different receptor tyrosine kinases came to similar conclusions. These studies contributed to the realization that tyrosine phosphorylation of proteins governs protein-protein interaction.
Kazlauskas A, Cooper JA. Autophosphorylation of the PDGF receptor in the kinase insert region regulates interactions with cell proteins. Cell 1989; 58:1121-33.
Kazlauskas A, Ellis C, Pawson T, Cooper JA. Binding of GAP to activated PDGF receptors. Science 1990; 247:1578-81.
Kazlauskas A, Cooper JA. Phosphorylation of the PDGF receptor β subunit creates a tight binding site for phosphatidylinositol 3 kinase. EMBO J 1990; 9:3279-86.
Valius M, Bazenet C, Kazlauskas A. Tyrosine 1021 and 1009 are phosphorylation sites in the carboxyterminus of the platelet-derived growth factor receptor β subunit and are required for binding of phospholipase Cγ and a 64 kd protein, respectively. Mol Cell Biol 1993; 13:133-43. -
Details
One of the key scientific questions at this point in time was how growth factors trigger cellular responses. The realization that SH2 domain-containing proteins that were being recruited to the activated PDGFR were signaling enzymes, and that their association with PDGFR often activated them, led to the hypothesis that such signaling enzymes were triggering signaling pathways that directed various cellular responses. Understanding how these signaling enzymes were being activated by growth factor receptors enabled the generation of reagents to selectively prevent the activation of a given signaling enzyme. These tools included PDGFR phosphorylation site mutants that selectively failed to engage a desired signaling enzyme. Working with a number of collaborators we defined the signaling enzymes that were essential for growth factor-driven proliferation and migration of cells. This information guided subsequent efforts to identify additional members of relevant signaling pathways, e.g. enzymes such as Akt, which acted downstream of phosphatidylinositol 3-kinase (PI3K).
Valius M, Kazlauskas A. Phospholipase Cγ and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor’s mitogenic signal. Cell 1993; 73:321-34.
Kundra V, Escobedo J, Kazlauskas A, Williams LT, Zetter B. Regulation of chemotaxis by the platelet-derived growth factor receptor-b. Nature 1994; 367:474-6.
Franke TF, Yang S-I, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the platelet-derived growth factor (PDGF)-activated phosphatidylinositol 3-kinase (PI 3-kinase). Cell 1995; 81:727-36.
Klinghoffer RA, Duckworth B, Valius M, Cantley L, Kazlauskas A. Platelet-derived growth factor-dependent activation of phosphatidylinositol 3-kinase is regulated by receptor binding of SH2-domain-containing proteins which influence Ras activity. Mol Cell Biol 1996; 16:5905-14. -
Details
At this point in time it was known that growth factors were capable of driving cells through the cell cycle, and that their input was required only for part of the cycle, namely, to move cells through the restriction point, which was close to the G1/S boundary. Which signaling enzymes were necessary, and when they contributed during this roughly 10-12 hr period was largely unknown. We discovered that growth factor-driven cell cycle progression did not require continuous signaling. Rather, growth factor-induced signaling was discontinuous, and appropriately timed discontinuous signaling was sufficient for progression through the cell cycle. We also identified which signaling enzymes were required and when they contributed. These discoveries guided subsequent investigation by other groups who defined the existence and timing of additional gating mechanisms for growth factor-driven cell cycle progression.
Jones SM, Klinghoffer R, Prestwich GD, Toker A, Kazlauskas A. PDGF induces an early and a late wave of PI 3-kinase activity, and only the late wave is required for progression through G1. Cur Biol 1999; 9:512-21.
Balciunaite E, Jones S, Toker A, Kazlauskas A. PDGF initiates two distinct phases of protein kinase C activity that make unequal contributions to the G0 to S transition. Cur Biol 2000; 10:261-7.
Jones SM, Kazlauskas A. Growth factor-dependent mitogenesis requires two distinct phases of signalling. Nature Cell Biol 2001; 3:165-72.
Balciunaite E, Kazlauskas A. Early phosphoinositide 3-kinase activity is required for late activation of protein kinase C epsilon in platelet-derived growth factor-stimulated cells: evidence for signaling across a large temporal gap. Biochem J 2001; 358:281-5. -
Details
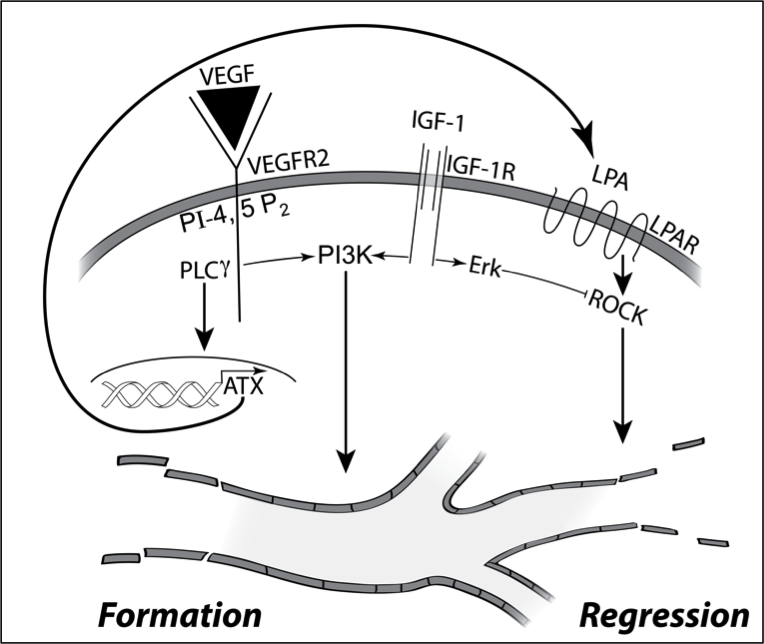
Focusing on the fate of newly formed blood vessels, we discovered that they are instructed to regress by the bioactive phospholipid LPA (lysophosphatidic acid). Since LPA is ubiquitous, how do newly formed blood vessels persist? The reason in part is because pro-angiogenic growth factors such as IGF-1 persistently activate signaling pathways that overcome LPA-directed regression. These concepts begin to explain why there are some many growth factors that participate in angiogenesis. Agents like VEGF trigger the formation of new blood vessel, while IGF-1 allows them to persist by overcoming the action of regression factors such as LPA.
These insights provided the foundation to investigate how diabetes disrupts angiogenic homeostasis. The high glucose aspect of diabetes rewires the signaling network in endothelial cells such that they become resistant to LPA-driven regression. This discovery begins to explain why pathological blood vessels accumulate in vitreous of patients with PDR (proliferative diabetic retinopathy), despite the presence of sufficient quantities of LPA to trigger their regression. Our contribution in this area has been both identification of signaling events that govern angiogenic homeostasis, and how diabetes rewires this network in endothelial cells.
Im E, Motiejunaite R, Aranda J, Park EY, Federico L, Kim TI, Clair T, Stracke ML, Smyth S, Kazlauskas A. Phospholipase Cgamma activation drives increased production of autotaxin in endothelial cells and lysophosphatidic acid-dependent regression. Mol Cell Biol 2010; 30:2401-10.
Aranda J, Motiejunaite R, Im E, Kazlauskas A. Diabetes Disrupts the Response of Retinal Endothelial Cells to the Angiomodulator Lysophosphatidic Acid. Diabetes 2012; 61:1225-33.
Motiejūnaitė R, Aranda J, Kazlauskas A. Pericytes prevent regression of endothelial cell tubes by accelerating metabolism of lysophosphatidic acid. Microvasc Res. 2014; 93:62-71.
Jacobo SM, Kazlauskas A. Insulin-like growth factor 1 (IGF-1) stabilizes nascent blood vessels. J Biol Chem. 2015; 290:6349-60. -
Details
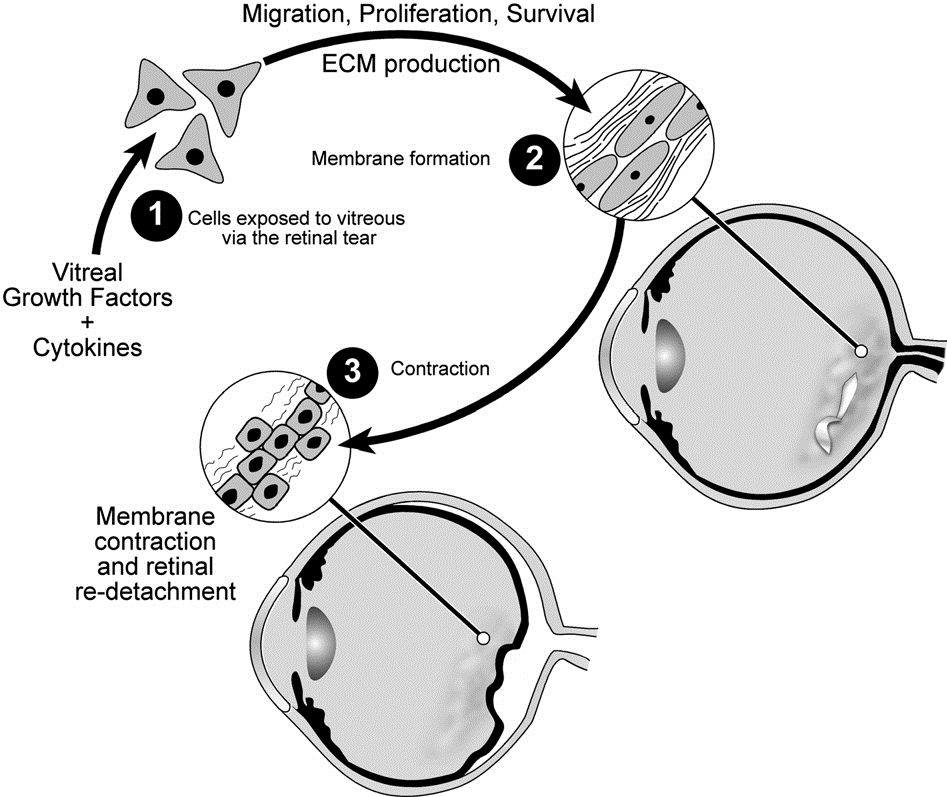
My most translational contribution to science has been in the area of a blinding disease called proliferative vitreoretinopathy (PVR). Patients who undergo surgery to correct a detached retina can develop this condition, which is difficult to manage. PVR develops when cells that are displaced into the vitreous proliferate, produce extracellular matrix and assemble it into an epiretinal membrane. Contraction of the epiretinal membrane detaches the retina and mandates additional episodes of surgery to re-attach the retina. We discovered that growth factors in the vitreous enable the survival of the misplaced cells. Furthermore, while there is a plethora of vitreal growth factors, their contribution to pathogenesis depends on their ability to indirectly activate PDGFRa, which is a novel mode of activation that promotes viability of displaced cells. Surprisingly, some of the growth factors antagonize the ability of other growth factors to indirectly activate PDGFRa. The discovery of both the hierarchy amongst vitreal growth factors, and the essential signaling events downstream of indirectly activated PDGFRa guided development of approaches to prevent experimental PVR.
Andrews A, Balciunaite E, Leong FL, Tallquist M, Soriano P, Refojo M, Kazlauskas A. Platelet-derived growth factor plays a key role in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci 1999; 40:2683-9.
Lei H, Velez G, Kazlauskas A. Pathological signaling via PDGFR{alpha} involves chronic activation of Akt and suppression of p53. Mol Cell Biol 2011; 31:1788-99.
Lei H, Kazlauskas A. A reactive oxygen species-mediated, self-perpetuating loop persistently activates platelet-derived growth factor receptor α. Mol Cell Biol. 2014; 34:110-22.
Pennock S, Rheaume MA, Mukai S, Kazlauskas A. A novel strategy to develop therapeutic approaches to prevent proliferative vitreoretinopathy. Am J Pathol 2011; 179:2931-40. -
Details
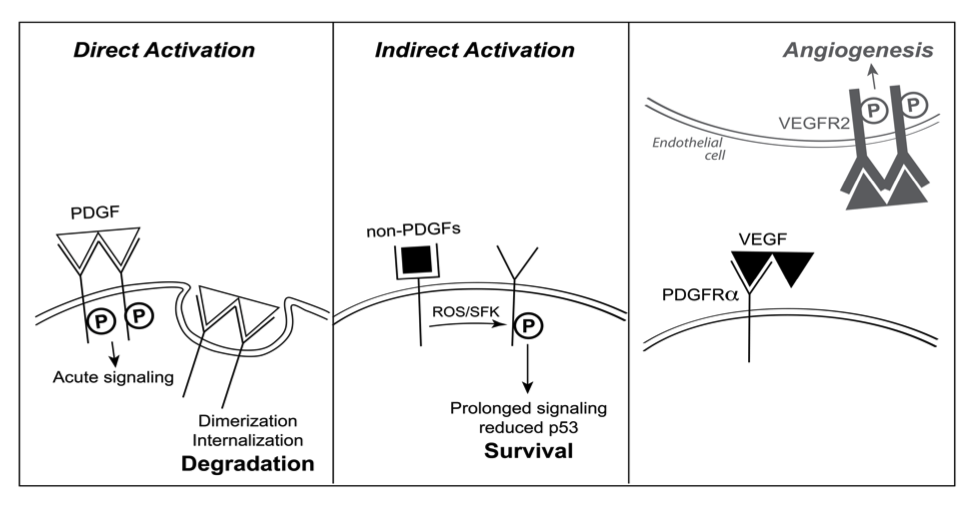
In the course of investigating the pathogenesis of PVR, we learned that three different classes of growth factors engage PDGFRa, and that there was a hierarchy amongst them. Growth factors outside of the PDGF family (non-PDGFs, such as EGF, bFGF, IGF-1, etc.) indirectly activated monomeric PDGFRa. The direct (PDGF-mediated) and indirect modes of activating PDGFRa were incompatible. In the presence of both PDGFs and non-PDGFs, the direct mode dominated because PDGF assembled PDGFRa into dimers, which were poorly activated by the indirect mode. While the direct mode of activation predominated when non-PDGFs and PDGF were present, the addition of VEGF-A switched the mode of activation to indirect. This is because VEGF-A, which has a very similar crystal structure to PDGF-B, compete with PDGF for binding to PDGFRa and thereby sustains a population of monomeric PDGFRas in the face of PDGF. The relevance of this newly appreciated, indirect mode of activating PDGFRa relates to its ability to reduce p53 and thereby promote survival of cells. In so doing, indirectly activated PDGFRa can either promote pathology (in the case of PVR), or physiology (by enhancing survival of healthy tissue during periods of hypoxia)
Lei H, Kazlauskas A. Growth factors outside of the PDGF family employ ROS/SFKs to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J Biol Chem 2009; 284:6329-36.
Pennock S, Kazlauskas A. Vascular endothelial growth factor A competitively inhibits platelet-derived growth factor (PDGF)-dependent activation of PDGF receptor and subsequent signaling events and cellular responses. Mol Cell Biol 2012; 32:1955-66.
Pennock S, Haddock LJ, Mukai S, Kazlauskas A. Vascular Endothelial Growth Factor Acts Primarily via Platelet-Derived Growth Factor Receptor α to Promote Proliferative Vitreoretinopathy. Am J Pathol. 2014; 184: 3052-68.
Pennock S, Kim, L, Kazlauskas A. VEGF-A acts via PDGFRα to promote viability of cells enduring hypoxia. Mol Cell Biol. 2016 Aug 26;36(18):2314-27.








